ę║ŽÓ│ŻęŖĄ─ē║┴”å¢Ņ}Ż¼╗∙ŠĆå¢Ņ}Ż¼ĘÕą╬å¢Ņ}Ą╚Č╝ėąĮŌ┤Ż¼ąĪ╗’░ķéā┌sŠoīżšę┤░Ė░╔ĪŻįÆ▓╗ČÓšfŻ¼ų▒Įė╔ŽĖ╔žøĪŻ║╬ų^Ę┤ŽÓų∙Ī󚲎Óų∙Ż┐
┤Ż║Ī░Ę┤ŽÓĪ▒║═Ī░š²ŽÓĪ▒Ą─Ė┼─Ņ╩Ūę║ŽÓ╔½ūVĘ©įńŲ┌╠ß│÷Ą─Ė┼─ŅŻ¼«öĢrµI║ŽŽÓ╔½ūVų∙╔ą╬┤│÷¼FŻ¼╣╠Č©ŽÓ▒╗═┐Ė▓į┌▌d¾w▒Ē├µŻ¼śOęū┴„╩¦Ż¼×ķ┤╦┐ŲīW╝ęī”┴„äėŽÓ╩╣ė├Įo│÷┴╦║Ž└ĒĄ─Į©ūhŻ║┴„äėŽÓśOąį┼c╣╠Č©ę║śOąį權▀ėą▌^┤¾▓ŅäeŻ¼ęį£p╔┘╣╠Č©ę║┴„╩¦ĪŻ╣╠Č©ŽÓśOąį╚§ė┌┴„äėŽÓĢrĄ─ę║ŽÓ╔½ūVĘ©▒╗ĘQ×ķĘ┤ŽÓ╔½ūVĘ©Ż¼╣╠Č©ŽÓśOąįÅŖė┌┴„äėŽÓĢrĄ─ę║ŽÓ╔½ūVĘ©▒╗ĘQ×ķš²ŽÓ╔½ūVĘ©ĪŻ▒M╣▄─┐Ū░µI║ŽŽÓ╔½ūVų∙ęč│╔×ķų„┴„Ż¼Ą½▀@ę╗Ė┼─Ņį┌╔½ūVĘĮĘ©ķ_░lĪóŅA£y│÷ĘÕĒśą“Ą╚ĘĮ├µŠ▀ėąųžę¬ęŌ┴xĪŻ
ė╔╔Ž├µĄ─ĮķĮB┐╔ų¬Š▀¾wĄ─╔½ūVĘĮĘ©Īó╔½ūVų∙ī┘ė┌š²ŽÓ▀Ć╩ŪĘ┤ŽÓ▓╗āH╚ĪøQė┌╣╠Č©ŽÓśOąįŻ¼═¼Ģr▀Ć╚ĪøQė┌┴„äėŽÓśOąįĪŻC18(╣Ķ─zµI║Ž╩«░╦═ķ╗∙╣Ķ═ķ)ĪóC8(╣Ķ─zµI║Žą┴╗∙╣Ķ═ķ)ĪóPH(╣Ķ─zµI║Ž▒Į╗∙╣Ķ═ķ)Ą╚╔½ūVų∙Ż¼ė╔ė┌╣╠Č©ŽÓśOąįśOĄ═Ż¼▒╚─┐Ū░ęčų¬Ą─╚╬║╬┴„äėŽÓĄ─śOąįČ╝ꬥ═Ż¼ę“Č°╩Ūś╦£╩Ą─Ę┤ŽÓų∙Ż╗Silica(╣Ķ─z)ĪóNH2(╣Ķ─zµI║Ž░▒▒¹╗∙╣Ķ═ķ)Š▀ėą▌^Ė▀Ą─śOąįŻ¼ų„ę¬ė├ė┌ĘųļxĦėąśOąį╗∙łFĄ─╗»║Ž╬’Ż¼╦∙ė├┴„äėŽÓĄ─śOąį═©│ŻĄ═ė┌▀@ą®╣╠Č©ŽÓŻ¼ę“Č°╩Ūś╦£╩Ą─š²ŽÓų∙ĪŻCN(╣Ķ─zµI║Žļµ▒¹╗∙)Ą─śOąį▀mųąŻ¼«ö┴„äėŽÓśOąį│¼▀^CNĢrŻ¼╦³ī┘ė┌Ę┤ŽÓų∙Ż¼Ę┤ų«ät╩Ūš²ŽÓų∙ĪŻ╔½ūVų∙ęÄĖ±ī”Ęų╬÷ĮY╣¹Ģ■«a╔·║╬ĘNė░ĒæŻ┐
┤Ż║╔½ūVų∙ā╚ÅĮøQČ©▌dśė┴┐Ż¼▌dśė┴┐┼cā╚ÅĮĄ─ŲĮĘĮ│╔š²▒╚Ż╗╔½ūVų∙ķLČ╚┼c╦■░ÕöĄ│╔š²▒╚Ż¼┼cų∙ē║│╔š²▒╚Ż╗┴ŻÅĮė░Ēæ£u┴„öU╔óŽÓŻ¼┴ŻÅĮįĮąĪ£u┴„öU╔óŽÓįĮąĪŻ¼ų∙ą¦įĮĖ▀Ż¼┴ŻÅĮ┼cų∙ą¦Į³╦Ų│╔Ę┤▒╚Ż╗┴ŻÅĮįĮąĪŻ¼ē║┴”ę▓įĮ┤¾Ż¼ē║┴”┼c┴ŻÅĮĄ─ŲĮĘĮ│╔Ę┤▒╚ĪŻ╠Ņ┴Ž┐ūÅĮī”Ęų╬÷ī”Ž¾Ą─Ęųūė┴┐ėąŽ▐ųŲŻ¼«ö┐ūÅĮ×ķĘų╬÷╬’│▀┤ńĄ─5▒Čęį╔ŽĢrŻ¼Ęų╬÷╬’▓┼─▄Ēś└¹═©▀^┐ūŽČŻ¼┐ūÅĮ╠Äė┌60~120 ÅĄ─╔½ūVų∙▀mė├ė┌ŽÓī”Ęųūė┴┐ąĪė┌10000Ą─Ęų╬÷╬’Ż¼┐ūÅĮ×ķ300 ÅĄ─╔½ūVų∙┐╔ęįØMūŃĘųūė┴┐╠Äė┌10000ęį╔ŽĄ─┤¾Ęųūė╗»║Ž╬’Ęų╬÷ĪŻę║ŽÓ╔½ūVĘų╬÷ųą╚ń║╬▓┼─▄╠ßĖ▀ĘųļxČ╚Ż┐┤Ż║Ž┬╩Į×ķĘųļxČ╚ėŗ╦Ń╣½╩Į
NŻ║ų∙ą¦(Efficiency)Ę┤ė│╔½ūVų∙ąį─▄Ż¼ų∙ą¦įĮĖ▀Ż¼ĘųļxČ╚įĮ║├ĪŻį┌Ųõ╦¹Śl╝■║ŃČ©Ą─ŪķørŽ┬Ż¼╦■░ÕöĄį÷╝ėę╗▒ČŻ¼ĘųļxČ╚āH╠ßĖ▀40%ĪŻ▓┘ū„ųąŻ¼┐╔═©▀^Ž┬├µā╔ĘNĘĮ╩Įį÷╝ė╦■░ÕöĄ▀MČ°╠ßĖ▀ĘųļxČ╚Ż║Ųõę╗Ż¼╩╣ė├ķLų∙╗“ļpų∙┤«┬ōŻ¼Ą½ę▓Ģ■╩╣ĘųļxĢrķg┤¾┤¾čėķLŻ╗ŲõČ■Ż¼╩╣ė├╝Ü┴ŻÅĮ╠Ņ┴ŽĄ─╔½ūVų∙Ż¼Ą½▀@ąĶę¬─═Ė³Ė▀ē║┴”Ą─ę║ŽÓ╔½ūVŽĄĮyĪŻŽÓ▒╚ų«Ž┬║¾š▀Ė³×ķ┐╔╚ĪĪŻ
”┴Ż║▀xō±ąį(selectivity)╩ŪųĖ╔½ūVų∙-┴„äėŽÓ¾wŽĄĘųļxā╔éĆ╗»║Ž╬’Ą──▄┴”ĪŻ▀xō±ąįų„ę¬┼c╣╠Č©ŽÓĪó┴„äėŽÓĮM│╔ęį╝░ų∙£žĄ╚ę“╦žėąĻPŻ¼┼c▒Ż┴¶ųĄę▓├▄ŪąŽÓĻPŻ¼Ųõųą╣╠Č©ŽÓ║═┴„äėŽÓĮM│╔ė░Ēæ▌^┤¾ĪŻęįūŅ│ŻęŖĄ─Ę┤ŽÓ─Ż╩Į×ķ└²Ż¼Ę┤ŽÓų∙(░³└©C18ĪóC8ĪóPHĄ╚)╩ŪęįĘų┼õū„ė├ī”╗»║Ž╬’▀Mąą▒Ż┴¶Ą─Ż¼▓╗═¼╗»║Ž╬’Ą─Ęųļx╩Ū╗∙ė┌╦³éāį┌µI║ŽŽÓ┼c┴„äėŽÓųąĘų┼õŽĄöĄĄ─▓Ņ«ÉŻ¼╚ń╣¹ā╔ĘN╗»║Ž╬’Ą─╦«╚▄ąįĪóį┌═ķ¤N-╦«¾wŽĄĄ─Ęų┼õŽĄöĄĄ╚ĘĮ├µ┤µį┌├„’@▓Ņ«ÉŻ¼─Ū├┤▀@ą®╗»║Ž╬’═©│Ż╩Ū─▄ē“└¹ė├Ę┤ŽÓų∙▀_ĄĮĘųļxŻ╗PHų∙ī”Š▀ėą▒ĮŁhĄ─╗»║Ž╬’Š▀ėą╠ž╩Ō▒Ż┴¶ĪŻ
š²ŽÓ─Ż╩ĮŽ┬Ż¼╣Ķ─zų∙Īó░Ę╗∙ų∙ĪóŪĶ╗∙ų∙┼cĦėąśOąį╗∙łFĄ─╗»║Ž╬’ų«ķg┤µį┌śOąįŽÓ╗źū„ė├Ż¼ī”╗»║Ž╬’Ą─╗∙łFŠ▀ėą▀xō±ąįŻ¼│Ż│Żė├ė┌ĮYśŗŅÉ╦Ų╬’Īó«Éśŗ¾w╗»║Ž╬’Ą─ĘųļxĪŻ┴„äėŽÓĘĮ├µŻ¼ĮĄĄ═┴„äėŽÓĄ─Ž┤├ōÅŖČ╚═©│Ż┐╔ęįį÷┤¾ĘųļxČ╚Ż╗Č°ėąÖC╚▄ä®ŅÉą═ę▓Ģ■ė░ĒæĘųļxŻ¼▒╚╚ńĘ┤ŽÓŚl╝■Ž┬Ż¼ęęļµ║═╝ū┤╝Ą─▀xō±ąįŠ═┤µį┌║▄┤¾▓Ņ«ÉŻ¼▀@ĘN▓Ņ«ÉąĶę¬į┌īŹ█`ųą├■╦„Ż¼Ą½¤ošō╚ń║╬Ż¼ČÓĘN╚▄ä®ŅÉą═ĦĮo╬ęéāĖ³ČÓĄ─īŹ¼FĘųļxĄ─┐╔─▄ĪŻ
kŻ║ļSų°╚▌┴┐ę“ūėkĄ─į÷┤¾Ż¼ĘųļxČ╚ę▓ļSų«į÷╝ėŻ¼▀@ĘNė░Ēæį┌kųĄ▌^Ą═ĢrĘŪ│Ż├„’@Ż¼«ökųĄ┤¾ė┌10ĢrŻ¼kųĄį÷╝ėī”ĘųļxČ╚Ą─ė░ĒæŠ═▓╗į┘’@ų°Ż¼▀@Š═Ėµš]¤oįŁätĄž╠ßĖ▀kųĄęįį÷┤¾ĘųļxČ╚╩Ūø]ėąęŌ┴xĄ─ĪŻį÷╝ėµI║ŽŽÓ├▄Č╚─▄ē“╠ßĖ▀kųĄŻ╗┴Ē═ŌĖ─ūāµI║Ž╗∙łFŅÉą═ę▓─▄Ė─ūākųĄŻ¼▒╚╚ńį┌Ę┤ŽÓ╔½ūVųąŻ¼ļSų°µI║ŽŽÓ╠╝µ£ķLČ╚Ą─į÷╝ėŻ¼kųĄųØuį÷┤¾ĪŻ╔½ūVĘÕĄ─ĘÕą╬╩Ūį§śė║Ō┴┐Ą─Ż┐ėą║╬ęÄČ©Ż┐└Ēšō╔ŽųvŻ¼╔½ūVĘÕæ¬Ę¹║ŽĖ▀╦╣Ū·ŠĆĘų▓╝Ż¼╚╗Č°īŹļH╔Ž╚╬║╬╔½ūVĘÕČ╝ī”Ė▀╦╣Ū·ŠĆĘų▓╝┤µį┌ę╗Č©Ą─Ų½ļxŻ¼ęÓ╝┤▓╗ī”ĘQĪŻĘÕ═Ž╬▓┐╔ęįė├▓╗ī”ĘQę“ūė(As)╗“USP═Ž╬▓ę“ūė(Tf)üĒ║Ō┴┐Ż¼’@╚╗▓╗ī”ĘQę“ūėĄ─šfĘ©Ė³£╩┤_Ż¼ę“×ķ╔½ūVĘÕ┤µį┌Ū░čėĪó═Ļ├└ī”ĘQĪó═Ž╬▓╚²ĘNą╬æBĪŻę╗░ŃüĒšfŻ¼ųŲ╦ÄąąśIęįUSP═Ž╬▓ę“ūėū„×ķįu£yś╦£╩Ż¼Č°Ųõ╦¹ąąśIätČÓ▓╔ė├AsüĒ£yČ©ĘÕą╬ĪŻ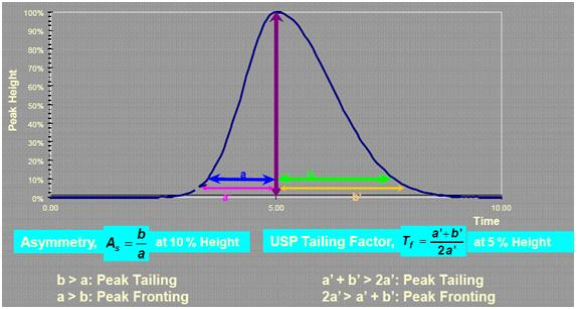
ī”ė┌╦Ä╬’Ęų╬÷Ż¼═©│Żėą├„┤_Ą─ęÄČ©Ż¼Tfæ¬╠Äė┌─│ę╗ĘČć·ų«ķgŻ¼▒╚╚ń╬ęć°╦ÄĄõęÄČ©─│ą®╦Ä╬’Ą─═Ž╬▓ę“ūėæ¬╠Äė┌0.95-1.05ų«ķgĪŻŲõ╦¹ąąśI╔ą¤o▌^×ķ├„┤_Ą─ęÄČ©ĪŻ╩▓├┤╩Ū╠▌Č╚Ž┤├ōŻ┐╩▓├┤ŪķørŽ┬╩╣ė├╠▌Č╚Ž┤├ōŻ┐
┤Ż║×ķ┴╦Ė─╔ŲĘų╬÷ĮY╣¹Ż¼─│ą®▓┘ū„ąĶę¬▀B└mĖ─ūā┴„äėŽÓųąĖ„╚▄ä®ĮMĘųĄ─▒╚└²ęį▀B└mĖ─ūā┴„äėŽÓĄ─śOąįŻ¼╩╣├┐éĆĘų╬÷ĮMĘųČ╝ėą║Ž▀mĄ─╚▌┴┐ę“ūėkŻ¼▓ó╩╣śėŲĘĘNĄ─╦∙ėąĮMĘų┐╔į┌ūŅČ╠Ģrķgā╚īŹ¼FūŅ╝čĘųļxŻ¼▀@ĘNŽ┤├ōĘĮ╩ĮĘQ×ķ╠▌Č╚Ž┤├ōĪŻ
╠▌Č╚Ž┤├ō┐╔į┌Ž┬┴ąŪķą╬ųą░lō]ųžę¬ū„ė├Ż║
A į┌Ą╚Č╚Ž┬Š▀ėą▌^īÆkųĄĄ─ČÓĘNśėŲĘĘų╬÷ĪŻ
B ┤¾ĘųūėśėŲĘĘų╬÷ĪŻ
C śėŲĘ║¼ėąÅŖ▒Ż┴¶Ą─Ė╔ö_╬’Ż¼į┌─┐ś╦╗»║Ž╬’│÷ĘÕ║¾įOų├╠▌Č╚Ž┤├ōŻ¼īóĖ╔ö_╬’Ž┤├ō│÷üĒŻ¼ęį├ŌŲõė░ĒæŽ┬ę╗┤╬öĄō■▓╔╝»ĪŻ
D Ęų╬÷ĘĮĘ©Į©┴óĢrŻ¼▓╗ų¬Ą└ŲõŽ┤├ōŪķørŻ¼╩╣ė├╠▌Č╚Ž┤├ōŻ¼šę│÷Ųõ▌^āץ─Ž┤├ōŚl╝■ĪŻ║╬ų^ĘŌČ╦Ż┐ĘŌČ╦Ą─ęŌ┴x╩Ū╩▓├┤Ż┐╣Ķ─z▒Ē├µĄ─╣Ķ┴u╗∙(Si-OH)├▄Č╚×ķ8 ”╠mol/m2Ż¼ė╔ė┌┐šķg╬╗ūĶĄ─┤µį┌Ż¼╣Ķ═ķ╗»µI║ŽĘ┤æ¬ūŅČÓų╗─▄Ė▓╔w50%Ą─╣Ķ┴u╗∙Ż¼│¼▀^ę╗░ļĄ─╣Ķ┴u╗∙╩Ū╗Ņąį╣Ķ┴u╗∙ĪŻ▀@ą®╣Ķ┴u╗∙┼c╗»║Ž╬’Ą─śOąį╗∙łF┤µį┌śOąįŽÓ╗źū„ė├║═ļxūėĮ╗ōQū„ė├Ż¼×ķ╗»║Ž╬’Ą─▒Ż┴¶į÷╝ė┴╦▓╗▒╗Ų┌═¹Ą─ū„ė├┴”Ż¼═∙═∙Ģ■ė░ĒæĘÕą╬ĪŻė├Č╠µ£┬╚╣Ķ═ķŻ©╚ń╚²╝ū╗∙┬╚╣Ķ═ķŻ®µI║Ž╗ŅąįĄ─╣Ķ┴u╗∙Ż¼┐╔ęį£pąĪ╔§ų┴Ž¹│²▀@ĘNė░ĒæŻ¼▀@ĘN▓┘ū„▒╗ĘQ×ķĘŌČ╦(Endcap)ĪŻ
ĘŌČ╦╠Ä└ĒĄ─ęŌ┴xŻ║
ęųųŲ┴╦╠ž«Éąį╬³ĖĮŻ¼╠ßĖ▀┴╦╔½ūVĘÕĄ─ī”ĘQąįŻ¼▓óĖ─╔Ų┴╦Ęųļxą¦╣¹Ż╗
į┌ę╗Č©│╠Č╚╔Žč┌▒╬┴╦╣Ķ─z▒Ē├µŻ¼╩╣Ųõī”ēAąįŁhŠ│Ą──═╩▄ąįį÷ÅŖŻ╗
═©▀^┐šķg╬╗ūĶč┌╔w┴╦µI║ŽĘ┤æ¬ą╬│╔Ą─Si-O-SiµIŻ¼╩╣Ųõī”╦ßąįŁhŠ│Ą──═╩▄ąįį÷ÅŖŻ╗┐╔─▄Ģ■ė░Ēæī”śOąįśėŲĘĄ─▀xō±ąįĪŻų∙┤▓╦·Ž▌║═µI║ŽŽÓ╦·Ž▌ų∙┤▓╦·Ž▌╩ŪųĖ╔½ūVų∙╩╣ė├ę╗Č╬Ģrķg║¾╔½ūVų∙╚ļ┐┌╠ÄĄ─ų∙┤▓«a╔·┐╔ęŖĄ─┐šŽČĪŻįō┐šŽČĄ─┤µį┌į÷┤¾┴╦╦└¾wĘeŻ¼Ģ■ī¦ų┬╔½ūVų∙ų∙ą¦Ž┬ĮĄĪŻįņ│╔ų∙┤▓╦·Ž▌Ą─įŁę“╚ńŽ┬Ż║
Ųõę╗Ż¼╔½ūVų∙╠ŅčbĢrĄ─ē║┴”▀^Ą═Ż¼╠Ņčb▓╗Šo├▄Ż¼į┌Ė▀ē║Ž┬╩╣ė├ę╗Č╬ĢrķgŻ¼ķ_╩╝│÷¼F┐šŽČŻ╗
ŲõČ■Ż¼▓┘ū„ē║┴”│¼│÷╔½ūVų∙╠Ņ┴ŽĄ──═ē║ųĄŻ¼ī¦ų┬╠Ņ┴ŽŅw┴ŻŲŲ╦ķ«a╔·┐šŽČŻ╗
Ųõ╚²Ż¼┴„äėŽÓ╚▄ĮŌ╠Ņ┴Žī¦ų┬┐šŽČ│÷¼FĪŻµI║ŽŽÓ╦·Ž▌╩ŪųĖė╔ė┌┴„äėŽÓśOąį┼cµI║ŽŽÓśOąįŽÓ▓Ņ╠½┤¾Ż¼µI║ŽŽÓ¤oĘ©į┌┴„äėŽÓųą│õĘų╔ņš╣Č°Ą╣Ę³Īó└pĮYį┌ę╗ŲŻ¼▒╚╚ńŲš═©C18į┌╝ā╦«ŽÓŚl╝■Ž┬ĪŻŽÓ╦·Ž▌Ģ■ī¦ų┬╔½ūVų∙ī”╗»║Ž╬’Ą─▒Ż┴¶▓╗ūŃĪŻŽĄĮyē║┴”╔²Ė▀Ą─įŁę“╝░┼┼│²╩╣ė├š▀═©▀^ė^▓ņāxŲ„Ą─ŽĄĮy▒O£yšŲ╬šŽĄĮyĄ─ē║┴”Ż¼╚ń╣¹░l¼Fē║┴”Ų½Ė▀Ż¼▓╗ę¬┴ó╝┤šJ×ķĪ░ų∙ē║Ė▀┴╦Ī▒Ż¼ę“×ķŽĄĮyē║┴”═©│Żė╔ų∙Ū░ē║┴”Īó╔½ūVų∙ē║┴”║═┴„═©│žē║┴”╝ė║ŽČ°│╔ĪŻ┤╦Ģrš²┤_Ą─ū÷Ę©╩ŪŻ║į┌▓┘ū„Śl╝■Ž┬Ż¼£yČ©ŽĄĮyē║┴”Ż¼Ą├ĄĮp┐éŻ╗ąČŽ┬╔½ūVų∙Ż¼į┌ŽÓ═¼Śl╝■Ž┬Ż¼£yČ©ų∙Ū░ē║┴”Ż¼Ą├ĄĮpŪ░Ż╗ė├ā╔═©īó▒├┴„│÷╣▄┬Ę┼c┴„═©│ž▀BĮėŻ¼į┌▓┘ū„Śl╝■Ž┬Ż¼£yČ©ē║┴”Ż¼Ą├ĄĮp(Ū░+║¾)ĪŻ═©▀^ėŗ╦Ńšę│÷ē║┴”Ė▀╩ŪüĒūįų∙Ū░Īó╔½ūVų∙▀Ć╩Ūų∙║¾ĪŻ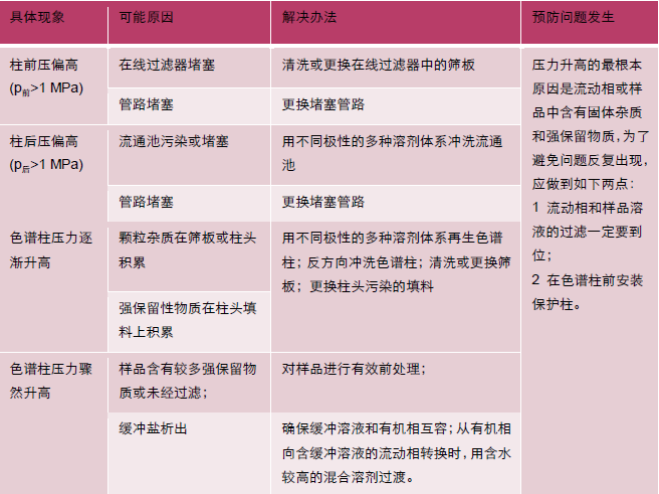
ęį╔ŽŪķą╬╝┘įOø]ėą░▓čb▒Żūoų∙Ż¼╚ń╣¹░▓čb┴╦▒Żūoų∙Ż¼ē║┴”╔²Ė▀Ģr欎╚£yįć▒Żūoų∙Ą─ē║┴”Ż¼ęį▒Ń┤_šJå¢Ņ}╩ŪʱüĒūį▒Żūoų∙ĪŻŽĄĮyē║┴”▓╗ĘĆČ©═©│ŻŪķørŽ┬Ż¼▒├ē║Ą─ūāäėųĄ│¼▀^┴╦0.5 MpaŻ¼ŽĄĮyē║┴”Š═ī┘ė┌▓╗ĘĆČ©Ą─ĘČ«ĀĪŻī¦ų┬ŽĄĮyē║┴”▓╗ĘĆČ©Ą─ūŅų▒ĮėįŁę“×ķ┴„äėŽÓ┴„┴┐║═ĮM│╔▌ö│÷Ą─▓╗ĘĆČ©Ż¼Č°ī¦ų┬┴„äėŽÓ▌ö│÷▓╗ĘĆČ©Ą─įŁę“═©│Ż░³└©╚▄ä®╗ź╚▌ąį▌^▓ŅĪóŽĄĮy┬®ę║ĪóŽĄĮy┤µį┌ÜŌ┼▌ęį╝░▌öę║ŽĄĮy▓┐╝■╣żū„╩¦ą¦ĪŻŽ┬├µīóĮķĮB┼┼│²Ī░ŽĄĮyē║┴”▓╗ĘĆĪ▒Ą─ĘĮĘ©ĪŻ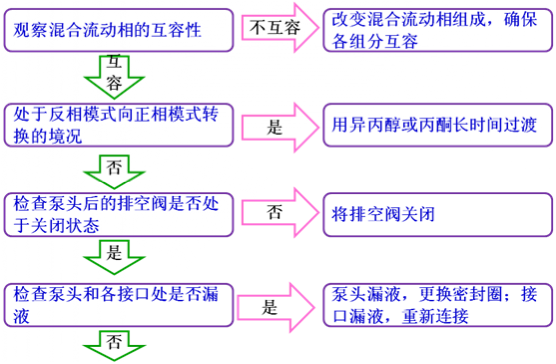
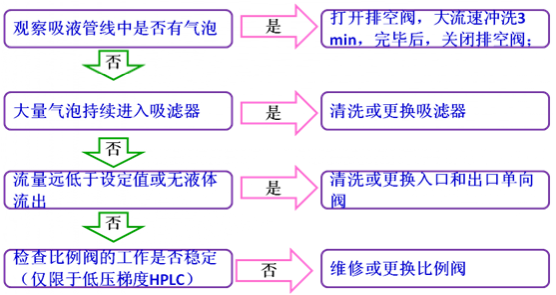
┴„äėŽÓ▌ö│÷▓╗ĘĆČ©▀ĆĢ■ī¦ų┬╗∙ŠĆ▓╗ĘĆČ©ęį╝░▒Ż┴¶Ģrķgūā╗»Ą╚¼FŽ¾Ż¼╦∙ęį╗∙ŠĆ▓╗ĘĆČ©╗“▒Ż┴¶Ģrķg▓╗ųž¼FĢrŻ¼Ž╚┐┤ę╗Ž┬ų∙ē║╩ŪʱĘĆČ©Ż¼╚ń╣¹▓╗ĘĆČ©Ż¼╚²éĆå¢Ņ}┐╔ęį║Ž▓ó╠Ä└ĒĪŻ╗∙ŠĆ▓╗ĘĆČ©
╗∙ŠĆ▓╗ĘĆČ©╩ŪųĖ╔½ūVŪ·ŠĆį┌▌^ķLĢrķgĘČć·ā╚ī”Ēææ¬ųĄĄ─0┐╠Č╚▌^┤¾Ą─Ų½ļxŻ¼═©│Ż░³└©╗∙ŠĆŲ»ęŲ(å╬ĘĮŽ“)Īó╗∙ŠĆŲĘ³(ęÄ┬╔Ą─╔ŽŽ┬ŲĘ³)Īóģ^ė“īÆČ╚śO┤¾Ą─╔½ūVĘÕ╚²ĘNŪķør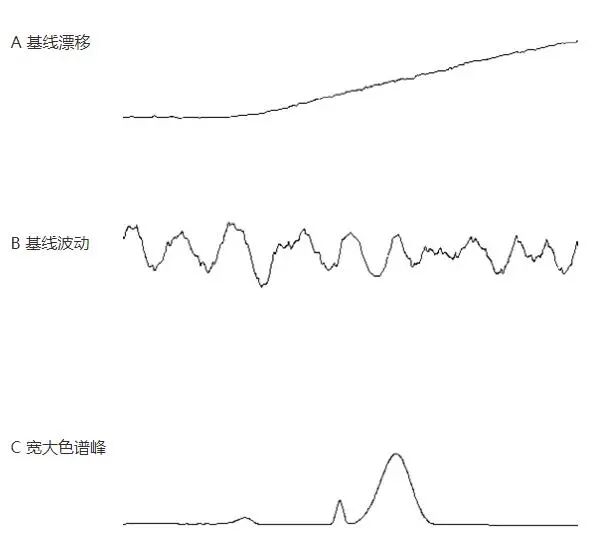
╚ń╣¹╗∙ŠĆ▓╗ĘĆČ©═¼Ģr░ķļSē║┴”▓©äėŻ¼šł░┤ššĪ░ē║┴”▓╗ĘĆČ©Ī▒▀Mąą┼┼▓ķĪŻ╚ń╣¹ē║┴”ĘĆČ©Ż¼šł░┤Ž┬▒ĒųąĄ─ĘĮĘ©▀Mąą┼┼▓ķĪŻ
įļ┬Ģ╔²Ė▀
įļ┬ĢŻ©baseline noiseŻ®Ż║ė╔Öz£yŲ„▌ö│÷┼c▒╗£yśėŲĘĮMĘų¤oĻPĄ─¤oęÄät▓©äėą┼╠¢Ż¼į┌╠žČ©ņ`├¶Č╚Ž┬ė├Ēææ¬å╬╬╗▒Ē╩ŠĪŻ┐╔Ęų×ķĖ▀Ņlįļ┬Ģ║═Č╠ų▄Ų┌įļ┬Ģā╔ĘNĪŻī”ė┌╣ŌūVą═Öz£yŲ„Ż¼įļ┬ĢĄ─š²│ŻĒææ¬ųĄ╠Äė┌0.000005 V ~ 0.00002 Vų«ķg(ģó┐╝Shimadzu Ą─HPLCāxŲ„)Ż¼Ė▀ė┌┤╦ųĄŻ¼╝┤┐╔šJ×ķįļ┬ĢŲ½Ė▀ĪŻ
įļ┬Ģ╔²Ė▀ĢrŻ¼Ž╚░┤Ž┬┴ą▓Į¾E┼┼▓ķ┴„äėŽÓå¢Ņ}
įļ┬Ģ╔²Ė▀ĢrŻ¼╚¶┴„äėŽÓø]ėąå¢Ņ}Ż¼æ¬┼┼▓ķāxŲ„Ą─å¢Ņ}
ų∙ąį─▄Ž┬ĮĄ
ų∙ąį─▄Ž┬ĮĄĄ─ę╗éĆųžę¬¼FŽ¾Š═╩Ūį┌▒Ż┴¶Ģrķg╗∙▒Š▓╗░l╔·ūā╗»Ą─ŪķørŽ┬Ż¼╔½ūVĘÕĄ─ģ^ė“īÆČ╚├„’@į÷┤¾(╚ńŽ┬łD)ĪŻ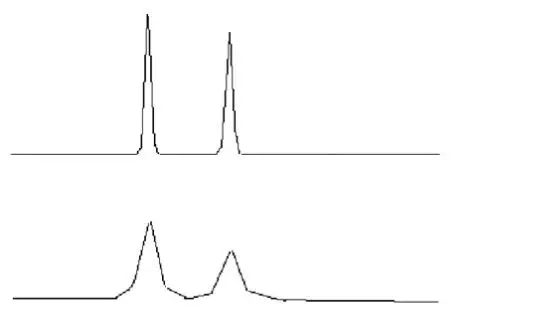
«öė÷ĄĮų∙ą¦Ž┬ĮĄŻ¼šł░┤Ž┬▒Ē▀Mąą┼┼▓ķ║═ĮŌøQ
▒Ż┴¶Ģrķgųž¼Fąį▓Ņ
«ö▒Ż┴¶ĢrķgĄ─Ų½▓Ņ│¼▀^2%Ż¼┐╔ęįšJ×ķ▒Ż┴¶ĢrķgĄ─ųž¼Fąį▓ŅĪŻī”ė┌─│ę╗éĆ╠žČ©Ą─Ęų╬÷Ż¼▒Ż┴¶Ģrķgų„ę¬╩▄┴„äėŽÓĮM│╔Īó┴„╦┘Īó£žČ╚ęį╝░╣╠Č©ŽÓė░ĒæŻ¼«ö▒Ż┴¶Ģrķg░l╔·├„’@ūā╗»ĢrŻ¼æ¬Å─▀@ÄūĘĮ├µ╚ļ╩ųĪŻ
A ė╔┴„äėŽÓįņ│╔Ą─▒Ż┴¶Ģrķg▓╗ĘĆČ©
B ė╔╣╠Č©ŽÓįņ│╔Ą─▒Ż┴¶Ģrķg▓╗ĘĆČ©
ĘÕą╬▓╗└ĒŽļ└ĒŽļĄ─╔½ūVĘÕū±čŁš²æBĘų▓╝Ż¼æ¬įōĘ¹║ŽĖ▀╦╣Ū·ŠĆŻ¼╩Ū═Ļ├└Č°ī”ĘQĄ─ĘÕą╬(╚ńłDA)Ż╗╚╗Č°ė╔ė┌╗»║Ž╬’ąį┘|Ą─╠ž╩ŌąįĪó╔½ūVų∙╣╩šŽĪó▓┘ū„Śl╝■Ą╚å¢Ņ}┤µį┌Ż¼ĘÕą╬┐╔─▄Ģ■▓╗ī”ĘQŻ¼Ū░čė(B)╗“═Ž╬▓(C)ĪŻ
ĘÕą╬Ū░čė(B)
ųą╦Ä│╔ĘųĘų╬÷ĒŚ─┐ųąŻ¼╠ß╚Ī╚▄ä®═©│Ż×ķęę┤╝Īó╝ū┤╝Īóęę╦ßę꧟Ą╚Ž┤├ōÅŖČ╚▌^ÅŖĄ─╚▄䮯¼┤╦Ģręū░l╔·Ī░śėŲĘ╚▄䮥─Ž┤├ōÅŖČ╚▀h▀h│¼▀^┴„äėŽÓĪ▒Ą─ŪķørŻ¼×ķ▒▄├ŌŪ░čėĘÕ│÷¼FŻ¼┐╔ęįė├═©▀^Ī░┴„äėŽÓŽĪßī╠ß╚Īę║Ī▒╗“ĮĄĄ═Ī░▀Mśė¾wĘeĪ▒Ą─ĘĮ╩ĮĪŻ
ĘÕą╬═Ž╬▓(C)
╣ĒĘÕ╣ĒĘÕ(Ghost peak)Ż║╩Ūī”╬┤ų¬üĒį┤Ą─╔½ūVĘÕĄ─ĮyĘQĪŻ
ÜŌ┼▌ĘÕÜŌ┼▌ĘÕ╩ŪųĖöyĦÜŌ┼▌Ą─┴„äėŽÓ═©▀^Öz£yŲ„ĢrĄ├ĄĮĄ─Ēææ¬ųĄŻ╗▀@└’Ą─ÜŌ┼▌┐╔─▄╩Ū┴„äėŽÓųąßīĘ┼Ą─┐šÜŌŻ¼ę▓┐╔─▄╩ŪėąÖCŽÓė÷╦«Ę┼¤ßČ°Ģ■ßīĘ┼Ą─ėąÖC╚▄ä®ĘųūėŻ╗ÜŌ┼▌ĘÕ═∙═∙Š▀ėą▌^Ė▀Ą─Ēææ¬ųĄŻ¼ģ^ė“īÆČ╚āH×ķöĄ├ļŻ¼│÷ĘÕĢrķg║═ŽÓæ¬ųĄ▒╚▌^ėąęÄ┬╔(╚ńŽ┬łD)ĪŻ═©│Żų╗ėą╣ŌūVą═Öz£yŲ„▓┼Ģ■│÷ÜŌ┼▌ĘÕĪŻ
ĮŌøQ▐kĘ©Ż║
├ōÜŌ
╩╣ė├ČÓį¬╠▌Č╚ĢrŻ¼ī”├┐ę╗ĘNĮMĘų▀Mąą├ōÜŌŻ¼┼┼│²╦³éā╚▄ĮŌĄ─┐šÜŌŻ╗
ī”ė┌ŅAŽ╚╗ņ║ŽĄ─╚▄䮯¼╗ņ║Ž║¾├ōÜŌŻ¼┼┼│²╚▄ĮŌĄ─┐šÜŌ║═ę“Ę┼¤ßČ°ō]░l│÷Ą─ėąÖC╚▄ä®ĘųūėŻ╗
Ū░├µĄ─├ōÜŌĘĮ╩Įī”ė┌ŅAŽ╚╗ņ║Ž║├Ą─╗ņ║Ž╚▄䮯¼╗∙▒Š┐╔ęį▀_ĄĮŽ¹│²ÜŌ┼▌ĘÕĄ──┐Ą─Ż╗Ą½ī”ė┌ČÓĘN╝ā╚▄ä®į┌āxŲ„ųą╗ņ║ŽĄ─ŪķørŻ¼╗ņ║Ž║¾╚▄䮾wŽĄĘ┼¤ßŻ¼┤µį┌Øōį┌Ą─ÜŌ┼▌Ż¼«ö┴„äėŽÓ┴„│÷╔½ūVų∙ĢrŻ¼ē║ÅŖ¾E£pŻ¼Øōį┌Ą─ÜŌ┼▌ęń│÷Ż¼▒╗Öz£yŲ„Öz£yĄĮĪŻ×ķ┴╦Ę└ų╣▀@ę╗å¢Ņ}│÷¼FŻ¼æ¬į┌Öz£yŲ„│÷┐┌▀BĮėę╗Č╬ā╚ÅĮ▌^╝ÜĄ─╣▄┬ĘŻ¼╩╣┴„äėŽÓ═©▀^Öz£yŲ„ĢrŻ¼ŁhŠ│ē║ÅŖ▌^Ė▀Ż¼ÜŌ┼▌ļyęįęń│÷Ż¼┐╔ęį▒▄├ŌÜŌ┼▌ĘÕ│÷¼FĪŻ
 ╩ųÖC░µ
╩ųÖC░µ




